Summary: Large circadian changes in blood pressure are possibly a very high risk factor for ischemic stroke. More data needed.
Interestingness: 2
Paper by Franz Halberg, Germaine Cornélissen, Julia Halberg, Henry Fink, Chen-Huan Chen, Kuniaki Otsuka, Yoshihiko Watanabe, Yuji Kumagai, Elena V. Syutkina, Terukazu Kawasaki, Keiko Uezono, Ziyan Zhao and Othild Schwartzkopff in the Journal of Anti-Aging Medicine, Volume 1, Issue 3, Fall 1998.
(((Back from hiatus. I found this paper more interesting than the usual, mainly because I hadn't heard about the topic before. Wikipedia calls the main author of this paper the founder of (American) chronobiology, and this paper seems to be part of the field. I'd never heard of it until now. The graph is full of what now would be considered retro-graphs which do help a lot)))
(((The language used in the paper is a bit salesmanish. It stresses two cases in which circadian hyper-amplitude-tension (CHAT) was diagnosed, with one case being treated, and the other not, and the large amount of money lost in treating the negative outcomes of the second case. I wouldn't be surprised if the field is considered quackish by academics)))
(((Switching back to paper mode))) The paper highlights the negative effects of having a high range (or double amplitude) (ie maximum value minus minimum value) in the smoothed measurements of blood pressure across the day. This is not about the difference between systolic and diastolic pressure but about comparing systolic vs systolic, or diastolic vs diastolic, throughout the day, and determining whether the differences are too high. The treatment recommended, briefly, consists of relaxation techniques and timed doses of anti-hypertension drugs.
To measure the double amplitude, a sine wave is fitted to the raw measurements which are taken across many days (((the more days the merrier it seems, but the ones mentioned seemed to fluctuate between 2 and 20 days))). (((least square error regression of the following formula:
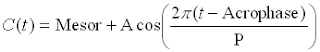
(image taken from
http://www.cbi.dongnocchi.it/glossary/Cosinor.html). The MESOR is the midline-estimating statistic of rhythm (some kind of mean), and the acrophase would be a phase adjustment. I think the MESOR, the amplitude, the period and the acrophase are fit simultaneously, but the period seems "seeded" to 24 hours))). Two separate curves are created, one for systolic and one for diastolic pressure. The MESOR is the value halfway between the peak and trough, and the double amplitude is the difference between the peak and trough of these curves. If the double amplitude measurement exceeds the 95th percentile for the person's particular age/gender bracket, that person is diagnosed with CHAT.
The main evidence presented as to the importance of CHAT is a study of 297 people who had their blood pressure monitored continuously for 48 hours, and then their incidence of negative vascular events recorded for six years (((Not sure what. Stroke and heart attacks I presume, but what else?))) The relative risks of the following conditions were calculated (Approximate 95% CI range in brackets):
- BMI > 25kg/m^2: 0.6 (0.2-2.1)
- High cholesterol: 1.0 (0.4-3)
- Male: 1.7 (0.55-4.8)
- Drinking: 2.5 (0.9-7.5)
- Family history: 2.6 (0.6-11)
- Smoking: 2.7 (1.0-8)
- Age > 60: 4.7 (1.6-12)
- Systolic MESOR > 130mmHg: 4.1 (1.0-16)
- Systolic CHAT: 6.2 (2.2-15)
- Diastolic CHAT: 8.2 (3-20)
(((The numbers of incidents were clearly quite low if the CI bars are so wide)))
Focusing on ischemic strokes, the relative risk of people with CHAT compared to people without CHAT are also much higher than 1.0 when partitioning the people into MESOR buckets, for every bucket, although in this case the CI ranges are even wider and include 1.0 in most cases. These high risk factors also remain when any of the other individual risk factors mentioned in the previous list are absent, and all with estimates higher than 5.0. Again, the ranges are big, but in this case, they do not touch 1.0.
Other studies cited are ones in which: 424 people that were measured for 24 hours, in which CHAT was related to higher left ventricular mass index; 18 11-14 year olds and the relation between CHAT and betamimetics received while in the womb; and a study of 40 rats in which CHAT preceded high MESOR by weeks.
Conclusion: More data would be nice to get so that the confidence interval ranges are tightened, and so that the findings don't feel like searching for the impressive statistic among a bunch of numbers. The relative risk values cited for stroke are impressive though. It could be a fun project over a week to check for CHAT.
Abstract follows:
Serial measurements, taken around the clock in the laboratory and clinic, can be analyzed by computer-implemented curve-fitting to assess the approximate 24-hour (circadian) variation, among other rhythmic and chaotic components of the time structure (chronome) of any variable. This approach is particularly important to quantify blood pressure variability, which renders even the most accurate single measurement into a snapshot on a roller coaster. A seemingly acceptable blood pressure can be particularly misleading when accompanied by the recommendation of another check-up in 2 years, which is the official position of the World Health Organization. An overswinging of the blood pressure along the 24-hour scale may then be missed. This excessive circadian amplitude, called "circadian hyper-amplitude-tension" (CHAT), constitutes a new disease risk syndrome, warranting screening, diagnosis, and treatment. With or without the midline-estimating statistic of rhythm (MESOR) (i.e., the [chronome-adjusted] mean value), the circadian double amplitude, a measure of the extent of predictable change within a day, is a predictor of vascular disease risk. An excessive amplitude (above the upper 95% prediction limit of healthy peers matched by age, gender, and ethnicity) is associated with an elevated left ventricular mass index in a retrospective chronometa-analysis of data from 424 patients and with an increase in morbid events in a prospective 6-year study on 297 patients, following-up on ancillary clinical studies and on results obtained on the laboratory model of the stroke-prone spontaneously hypertensive rat. CHAT is associated with a 720% increase in risk of ischemie cerebral events. It represents the greatest increase in risk, compared with 310%, 370%, 160%, 170%, and 150% in relation to a high blood pressure, old age, a family history of high blood pressure, and/or of other vascular disease, smoking and alcohol consumption, respectively. To identify CHAT and for other diagnostic and therapeutic reasons, single measurements should be replaced by an around-the-clock profile, for a week or longer, if need be, at the outset. The profile is preferably obtained by automatic monitoring with ambulatorily functional instrumentation. When such a monitor is unavailable, self-measurements at 3-hour intervals during waking and one around midsleep are acceptable. The midsleep measurement is taken with minimal disturbance, preferably by a companion, while the patient sleeps with a cuff on the arm. When no companion is available, the patient can set an alarm clock to take the self-measurement. Treatment should be timed with individualized guidance by a blood pressure profile (chronotherapy). The same profile also serves to assess the treatment effect with a control chart to validate the reduction of an excessive amplitude, the lowering of the blood pressure, or both when elevated. Controlled clinical trials assessing long-term outcomes are overdue. By monitoring for only weeks, the recognition and treatment of blood pressure overswinging along the 24-hour scale—a must in anti-aging medicine—may prevent postcatastrophic care for years.


